Tutorial 3: mouse somatosensory cortex by osmFISH
the osmFISH data is available at http://linnarssonlab.org/osmFISH/availability/
Loading and Preparing Data
import pandas as pd
import numpy as np
import scanpy as sc
import matplotlib.pyplot as plt
from sklearn.metrics import adjusted_rand_score
from sklearn.metrics.cluster import normalized_mutual_info_score as nmi
from SEPAR_model import SEPAR
# Read data
# adata = sc.read_h5ad(filename='osmfish_remove_excluded.h5ad')
filename = 'dataset/osmfish/osmfish_remove_excluded.h5ad'
adata = sc.read_h5ad(filename)
adata.var_names_make_unique()
# Get spatial coordinates
loc = adata.obsm['spatial']
# Get ground truth annotations
Y_list = adata.obs['Region']
n_cluster = len(Y_list.unique())
# Initialize SEPAR
separ = SEPAR(adata, n_cluster=n_cluster)
Data Preprocessing
# Compute spatial graph with custom radius
separ.compute_graph(radius_rate=1.1)
# Compute weights
separ.compute_weight(n_cluster=n_cluster)
1. Running SEPAR Algorithm
# Run SEPAR algorithm
separ.separ_algorithm(
r=30, # Number of spatial patterns
alpha=1.0, # Graph regularization weight
beta=0.05, # Sparsity penalty weight (previously l1)
gamma=0.01 # Pattern orthogonality weight (previously lam)
)
Processing iterations: 100%|██████████| 100/100 [00:00<00:00, 128.77it/s]
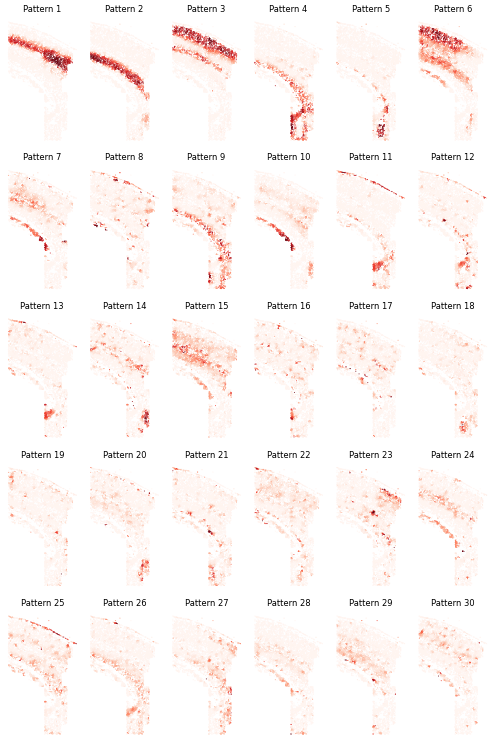
Pattern Visualization
# Calculate pattern significance
sim_slt = separ.sim_res(separ.Wpn, separ.Hpn, separ.Xt.T)
sim_argsort = np.argsort(-sim_slt)
# Visualize patterns
num_patterns = 30
plt.figure(dpi=50, figsize=(10, 15))
for i in range(num_patterns):
ii = sim_argsort[i]
plt.subplot(5, int(np.ceil(num_patterns/5)), i + 1)
plt.scatter(separ.loc[:, 0], separ.loc[:, 1],
c=separ.Wpn[:, ii].reshape(-1, 1),
s=0.2, cmap='Reds')
plt.axis('off')
plt.title(f'Pattern {i + 1}',
fontsize=12)
plt.tight_layout()
plt.show()
2. Performing Clustering and Evaluation
# Perform clustering
cluster_res = separ.clustering(
n_cluster=n_cluster,
N1=0,
N2=0
)
# Calculate clustering metrics
Ari = adjusted_rand_score(Y_list, separ.labelres)
Nmi = nmi(Y_list, separ.labelres)
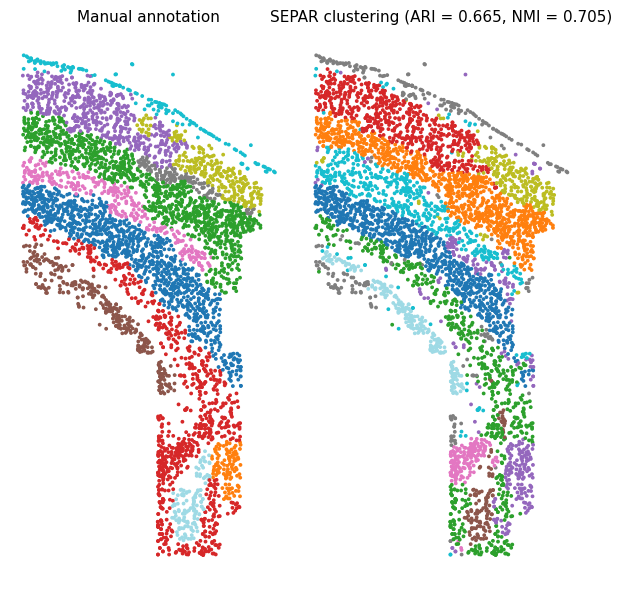
Clustering Results Visualization
# Create comparison visualization
fig, axs = plt.subplots(1, 2, figsize=(6, 6))
# Ground truth annotation
scatter = axs[0].scatter(separ.loc[:, 0], separ.loc[:, 1],
c=Y_list.map(lambda x: list(Y_list.unique()).index(x)),
s=3, cmap='tab20')
axs[0].set_title("Manual annotation", fontsize=11)
axs[0].axis('off')
# SEPAR clustering results
scatter = axs[1].scatter(separ.loc[:, 0], separ.loc[:, 1],
c=separ.labelres, s=3, cmap='tab20')
axs[1].set_title(f"SEPAR clustering (ARI = {Ari:.3f}, NMI = {Nmi:.3f})",
fontsize=11)
axs[1].axis('off')
plt.tight_layout()
plt.show()
3. Gene Expression Refinement
adata_refined = separ.get_refined_expression()
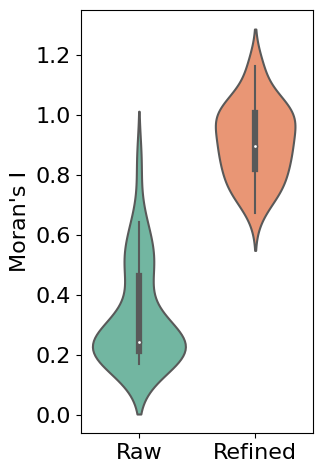
Evaluating Refinement Performance
# Calculate Moran's I for both raw and refined data
morani = sc.metrics.morans_i(separ.adata)
morani_refine = sc.metrics.morans_i(adata_refined)
# Create DataFrame for violin plot
data_violin = pd.DataFrame({
'Moran\'s I': np.concatenate([morani, morani_refine]),
'Condition': ['Raw'] * len(morani) + ['Refined'] * len(morani_refine)
})
import seaborn as sns
# Create violin plot
plt.rcParams['font.size'] = 16
plt.figure(figsize=(3, 5.5))
sns.violinplot(x='Condition', y='Moran\'s I', data=data_violin, palette='Set2')
plt.ylabel('Moran\'s I')
plt.xlabel('')
plt.show()
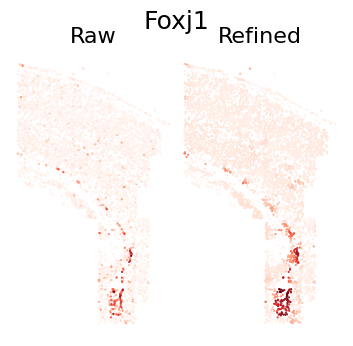
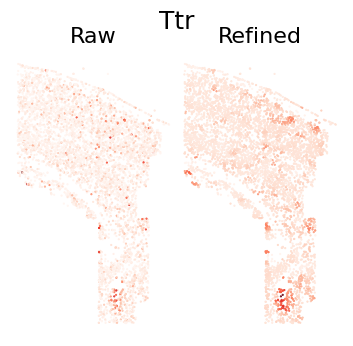
Single Gene Visualization
def plot_gene_refinement(gene_name, separ, adata_refined):
"""
Visualize raw and refined expression patterns for a specific gene.
Parameters
----------
gene_name : str
Name of the gene to visualize
separ : SEPAR object
SEPAR object containing raw data
adata_refined : AnnData
Refined expression data from get_refined_expression()
"""
# Get gene index and expression values
gene_idx = separ.adata.var_names.get_loc(gene_name)
raw_exp = separ.adata[:, gene_idx].X.toarray().flatten()
refined_exp = adata_refined.X[:, gene_idx]
# Create comparison plot
plt.rcParams['font.size'] = 16
plt.figure(dpi=100, figsize=(4/1.2, 4/1.2))
# Set main title
plt.suptitle(gene_name, fontsize=18)
# Plot raw expression
plt.subplot(1, 2, 1)
plt.scatter(separ.loc[:, 0], separ.loc[:, 1],
c=raw_exp, s=0.3, cmap='Reds', rasterized=True)
plt.axis('off')
plt.title('Raw', fontsize=16)
# Plot refined expression
plt.subplot(1, 2, 2)
plt.scatter(separ.loc[:, 0], separ.loc[:, 1],
c=refined_exp, s=0.3, cmap='Reds', rasterized=True)
plt.axis('off')
plt.title('Refined', fontsize=16)
plt.tight_layout(pad=0)
plt.show()
plot_gene_refinement("Foxj1", separ, adata_refined)
plot_gene_refinement("Ttr", separ, adata_refined)